Differential abundance analysis of proteomics data with pylimma
This tutorial demonstrates the AnnData-native pylimma workflow on a bulk mass-spectrometry proteomics dataset. The matrix is already log-transformed and median-normalised, so the analysis is a classical microarray-style limma pipeline: build a design matrix, fit per-protein linear models, moderate variances with empirical Bayes, and read off the top differentially abundant proteins.
Dataset
Mulvey et al. 2026, Mol Cell Proteomics 25(2):101510. doi:10.1016/j.mcpro.2026.101510. Tissue proteomics from human left-ventricular tissue collected at heart transplantation or LVAD implantation (heart-failure cases) or from organ donors (controls). The original paper compares peripartum cardiomyopathy hearts against non-peripartum cardiomyopathy and controls; for this tutorial we collapse the two cardiomyopathy groups into a single
heart_failure arm and contrast it against controls.
Shape: 7,026 protein groups x 15 samples (6 control donors + 9 heart-failure cases).
Pipeline
Load the protein abundance matrix and sample targets
Build an AnnData object (samples on
obs, proteins onvar)PCA QC
Design matrix:
~ grouplm_fit->e_bayesTop differentially abundant proteins (heart-failure vs control)
Volcano plot
Loading the data
The two CSVs live under pylimma/data/ alongside the other bundled benchmark datasets (ALL, GSE60450, pasilla, Yoruba); see pylimma/data/DATA_PROVENANCE.md for the full provenance and licence summary.
[1]:
import sys
import warnings
from pathlib import Path
import anndata as ad
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
warnings.filterwarnings('ignore', category=FutureWarning)
# This notebook lives at pylimma/pylimma/examples/mulvey/
# parents[1] = repo root (containing pylimma source + data/), matching
# the convention used by the other example notebooks.
REPO = Path.cwd().resolve().parents[1]
sys.path.insert(0, str(REPO))
import pylimma
DATA_DIR = REPO / 'data'
assert DATA_DIR.exists(), f'expected data dir at {DATA_DIR}'
pd.set_option('display.width', 120)
pd.set_option('display.max_columns', 12)
/Users/John/miniconda3/lib/python3.11/site-packages/h5py/__init__.py:36: UserWarning: h5py is running against HDF5 1.14.3 when it was built against 1.14.2, this may cause problems
_warn(("h5py is running against HDF5 {0} when it was built against {1}, "
1. Load the matrix and targets
[2]:
proteome = pd.read_csv(DATA_DIR / 'mulvey_proteome.csv.gz', index_col=0)
targets = pd.read_csv(DATA_DIR / 'mulvey_targets.csv').set_index('sample_id')
protein_meta_cols = ['gene_symbol', 'num_psm',
'max_pep_prob', 'reference_intensity']
sample_cols = [c for c in proteome.columns
if c not in protein_meta_cols]
X = proteome[sample_cols].T.values
var_df = proteome[protein_meta_cols].copy()
var_df.index.name = 'uniprot_id'
obs_df = targets.loc[sample_cols].copy()
obs_df['group'] = pd.Categorical(obs_df['group'],
categories=['control', 'heart_failure'],
ordered=False)
adata = ad.AnnData(X=X, obs=obs_df, var=var_df)
adata.obs_names = sample_cols
adata.var_names = var_df.index
print(f'AnnData shape: {adata.shape} (samples x proteins)')
print(f'\ngroup counts:\n{adata.obs.group.value_counts()}')
adata.var.head()
AnnData shape: (15, 7026) (samples x proteins)
group counts:
group
heart_failure 9
control 6
Name: count, dtype: int64
[2]:
| gene_symbol | num_psm | max_pep_prob | reference_intensity | |
|---|---|---|---|---|
| uniprot_id | ||||
| A0A075B6H7 | IGKV3-7 | 2 | 1.0000 | 16.264649 |
| A0A075B6H9 | IGLV4-69 | 2 | 1.0000 | 18.129858 |
| A0A075B6I0 | IGLV8-61 | 2 | 1.0000 | 18.895982 |
| A0A075B6I1 | IGLV4-60 | 3 | 1.0000 | 17.037598 |
| A0A075B6I4 | IGLV10-54 | 2 | 0.9998 | 15.915174 |
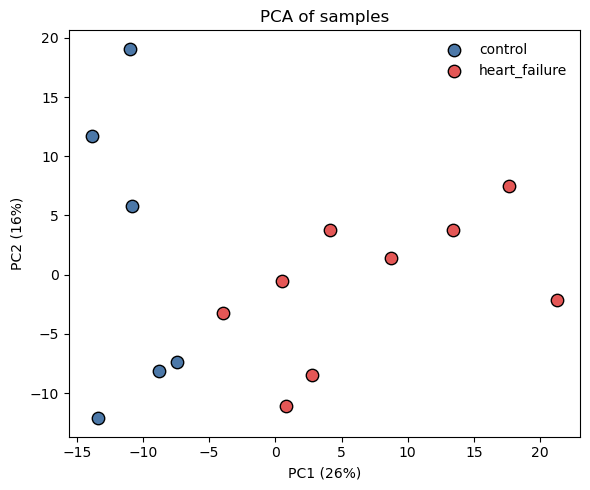
2. PCA QC
A PCA projection is a basic diagnostic to inspect before any DE analysis: separation between groups along an early PC indicates the contrast carries the expected signal.
[3]:
fig, ax = plt.subplots(figsize=(6, 5))
# PCA on samples (centre proteins, drop any all-NaN rows just in case).
Xc = adata.X - adata.X.mean(axis=0, keepdims=True)
U, s, Vt = np.linalg.svd(Xc, full_matrices=False)
pcs = U * s
var_explained = (s ** 2) / (s ** 2).sum()
group_palette = {'control': '#4c78a8', 'heart_failure': '#e45756'}
for grp, colour in group_palette.items():
m = (adata.obs['group'] == grp).values
ax.scatter(pcs[m, 0], pcs[m, 1], s=80,
color=colour, edgecolor='black', label=grp)
ax.set_xlabel(f'PC1 ({var_explained[0]:.0%})')
ax.set_ylabel(f'PC2 ({var_explained[1]:.0%})')
ax.set_title('PCA of samples')
ax.legend(frameon=False)
fig.tight_layout()
plt.show()
3. Design matrix and contrast
Two-level group factor with control as the reference level. The design has an intercept plus a heart_failure indicator; the contrast that asks “what is differentially abundant in heart failure versus control” is just the coefficient on the indicator (a one-element contrast vector [0, 1]).
[4]:
group = adata.obs['group']
design = pd.DataFrame({
'Intercept': 1.0,
'heart_failure': (group == 'heart_failure').astype(float).values,
}, index=adata.obs_names)
contrasts = pylimma.make_contrasts(HFvsCtrl='heart_failure', levels=design)
print('design matrix:')
print(design.head(3))
print('\ncontrast:')
print(contrasts)
design matrix:
Intercept heart_failure
Ctrl1 1.0 0.0
Ctrl2 1.0 0.0
Ctrl3 1.0 0.0
contrast:
HFvsCtrl
Intercept 0.0
heart_failure 1.0
4. lm_fit -> contrasts_fit -> e_bayes
Pure AnnData pipeline. The fit is stored in adata.uns['pylimma'] and survives write_h5ad if you want to round-trip it through disk. No voom step here: the input is already log-scale normalised abundance, so we feed it straight into lm_fit.
[5]:
pylimma.lm_fit(adata, design)
pylimma.contrasts_fit(adata, contrasts=contrasts)
pylimma.e_bayes(adata)
print('keys stored in adata.uns["pylimma"]:')
print(sorted(adata.uns['pylimma'].keys()))
keys stored in adata.uns["pylimma"]:
['Amean', 'F', 'F_p_value', 'coef_names', 'coefficients', 'contrast_names', 'contrasts', 'cov_coefficients', 'design', 'df_prior', 'df_residual', 'df_total', 'genes', 'lods', 'method', 'p_value', 'pivot', 'proportion', 'rank', 's2_post', 's2_prior', 'sigma', 'stdev_unscaled', 't', 'targets', 'var_prior']
5. Top differentially abundant proteins
[6]:
top = pylimma.top_table(adata, coef='HFvsCtrl',
number=np.inf, sort_by='p')
# Carry over gene symbols from adata.var so the table is readable.
top = top.join(adata.var[['gene_symbol']], how='left')
n_sig_05 = int((top['adj_p_value'] < 0.05).sum())
n_sig_01 = int((top['adj_p_value'] < 0.01).sum())
print(f'differentially abundant proteins at adj_p_value < 0.05: {n_sig_05:,}')
print(f'differentially abundant proteins at adj_p_value < 0.01: {n_sig_01:,}')
display_cols = ['gene_symbol', 'log_fc', 'ave_expr', 't',
'p_value', 'adj_p_value', 'b']
top.loc[:, display_cols].head(15).round(3)
differentially abundant proteins at adj_p_value < 0.05: 267
differentially abundant proteins at adj_p_value < 0.01: 0
[6]:
| gene_symbol | log_fc | ave_expr | t | p_value | adj_p_value | b | |
|---|---|---|---|---|---|---|---|
| gene | |||||||
| P02768 | ALB | 0.776 | 10.394 | 7.232 | 0.0 | 0.012 | 5.006 |
| O43272 | PRODH | -1.117 | 0.421 | -6.932 | 0.0 | 0.012 | 4.550 |
| Q9HDC5 | JPH1 | -0.522 | -0.142 | -6.422 | 0.0 | 0.015 | 3.738 |
| Q9UBX5 | FBLN5 | 1.266 | 2.883 | 6.364 | 0.0 | 0.015 | 3.644 |
| P0DJI8 | SAA1 | -1.665 | 0.410 | -6.290 | 0.0 | 0.015 | 3.522 |
| Q8WTS1 | ABHD5 | -0.533 | -0.134 | -6.077 | 0.0 | 0.015 | 3.166 |
| Q53HC5 | KLHL26 | 0.466 | -3.695 | 6.060 | 0.0 | 0.015 | 3.137 |
| P31939 | ATIC | -0.412 | 4.120 | -6.045 | 0.0 | 0.015 | 3.112 |
| Q6UX53 | METTL7B | -0.867 | 0.794 | -5.841 | 0.0 | 0.015 | 2.762 |
| L0R8F8 | MIEF1 | -0.513 | -1.684 | -5.834 | 0.0 | 0.015 | 2.751 |
| Q9H511 | KLHL31 | -0.392 | 1.800 | -5.796 | 0.0 | 0.015 | 2.685 |
| Q9UI14 | RABAC1 | -0.474 | -1.084 | -5.794 | 0.0 | 0.015 | 2.680 |
| O60437 | PPL | -0.678 | 0.905 | -5.767 | 0.0 | 0.015 | 2.634 |
| Q3ZCW2 | LGALSL | 0.448 | -0.162 | 5.751 | 0.0 | 0.015 | 2.607 |
| P05452 | CLEC3B | 0.809 | 1.326 | 5.477 | 0.0 | 0.022 | 2.123 |
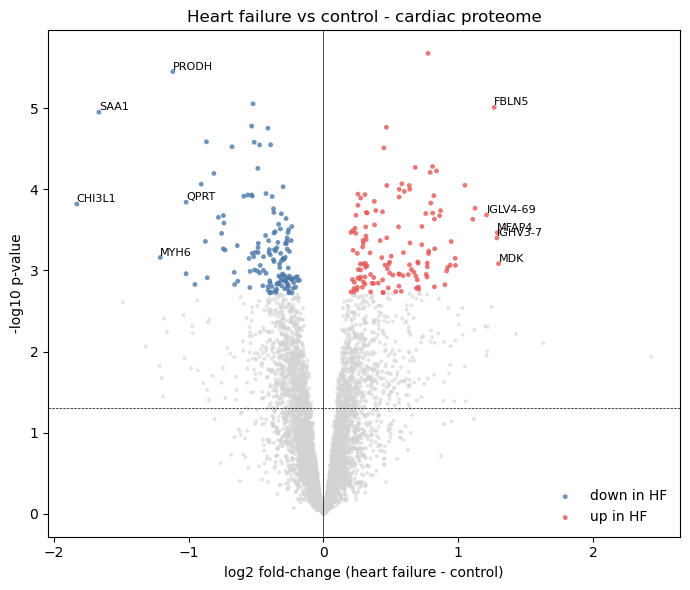
6. Volcano plot
Standard summary plot. Each point is one protein group; the x-axis is the log fold-change in heart failure relative to control, the y-axis is -log10(p_value). Proteins past adj_p_value < 0.05 are coloured red (up in heart failure) or blue (down in heart failure). Top hits by signed log fold-change are labelled with their gene symbol.
[7]:
fig, ax = plt.subplots(figsize=(7, 6))
sig_up = (top['adj_p_value'] < 0.05) & (top['log_fc'] > 0)
sig_down = (top['adj_p_value'] < 0.05) & (top['log_fc'] < 0)
ns = ~(sig_up | sig_down)
nlp = -np.log10(np.maximum(top['p_value'].values, 1e-300))
ax.scatter(top.loc[ns, 'log_fc'], nlp[ns.values],
s=8, color='lightgrey', alpha=0.6, edgecolor='none')
ax.scatter(top.loc[sig_down, 'log_fc'], nlp[sig_down.values],
s=12, color='#4c78a8', alpha=0.8,
edgecolor='none', label='down in HF')
ax.scatter(top.loc[sig_up, 'log_fc'], nlp[sig_up.values],
s=12, color='#e45756', alpha=0.8,
edgecolor='none', label='up in HF')
# Label the top 5 up and top 5 down by signed log fold change among the
# significant proteins (those past adj_p_value < 0.05). Gene symbols
# only - some entries have multi-gene proteingroup labels which we skip
# to keep the plot legible.
sig = top[top['adj_p_value'] < 0.05].copy()
top_up = sig.sort_values('log_fc', ascending=False).head(5)
top_down = sig.sort_values('log_fc', ascending=True ).head(5)
for _, row in pd.concat([top_up, top_down]).iterrows():
label = row['gene_symbol']
if not isinstance(label, str) or ';' in label or label == '':
continue
ax.annotate(label, (row['log_fc'], -np.log10(max(row['p_value'], 1e-300))),
fontsize=8, ha='left', va='bottom')
ax.axhline(-np.log10(0.05), color='black', lw=0.5, linestyle='--')
ax.axvline(0, color='black', lw=0.5)
ax.set_xlabel('log2 fold-change (heart failure - control)')
ax.set_ylabel('-log10 p-value')
ax.set_title('Heart failure vs control - cardiac proteome')
ax.legend(frameon=False, loc='lower right')
fig.tight_layout()
plt.show()